服务热线 : 400-025-7300



对于设计用于析氢反应(HER)的高效电催化剂,尚不清楚不昂贵的过渡金属作为单原子催化剂(SACs)的选择。本文中,报道了活性与催化剂、电子结构的相关性,以通过结合密度泛函理论计算和电化学测量来阐明负载在氮掺杂石墨烯上的一系列过渡金属作为SACs用于HER的反应性起源。只有极少数对HER表现出良好的催化活性,因为SACs的过渡金属(例如,Co、Cr、Fe、Rh和V)的吉布斯自由能在–0.20至0.30 eV的范围内变化,其中Co-SAC在0.13 eV时具有最高的电化学活性。电子结构研究表明,活性价dz2轨道的能量状态及其产生的反键状态决定了HER的催化活性。在Co-SAC的情况下,反键态轨道既不完全为空也不完全填充,这是其理想的氢吸附能的主要原因。此外,电化学测量表明,Co-SAC具有比Ni-SAC和W-SAC更好的析氢活性,从而证实了理论计算。这项系统的研究对高效SACs用于HER的设计提供了基本的了解。
.png)
Figure 1. 提出的单原子催化剂模型及其对氢吸附反应的吉布斯自由能计算:a)在金属官能化的N掺杂石墨烯中氢吸附的非金属位点;b)每个位点氢吸附反应的吉布斯自由能(ΔGH*);c)与石墨烯片中的四个氮原子配位的金属活性位点;d)一系列过渡金属用作单原子催化剂对氢吸附反应的吉布斯自由能(ΔGH*)图。
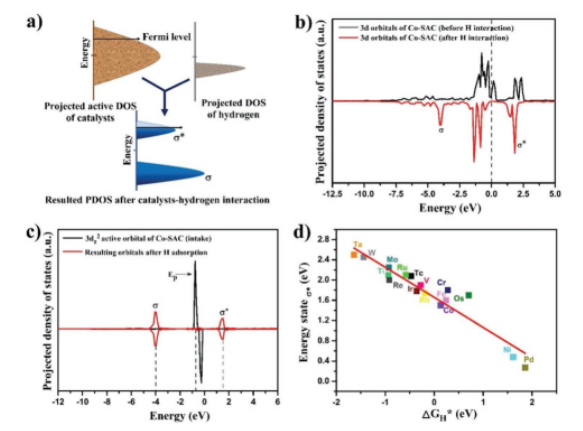
Figure 2. 单原子催化剂的电子结构及其活性关系:a)催化剂活性位点与氢的轨道杂交方案(σ=键,σ*=反键态轨道);b)Co-SAC氢相互作用前后3d杂化轨道的预计DOS;c)Co-SAC的3dz2活跃轨道(Ep,向上旋转和向下旋转)的PDOS与氢相互作用产生σ和σ*轨道;d)吉布斯自由能(ΔGH*)与反键态,费米能级上的Eσ*之间的相关性源自单原子催化剂的活性价dz2轨道与氢原子的相互作用。
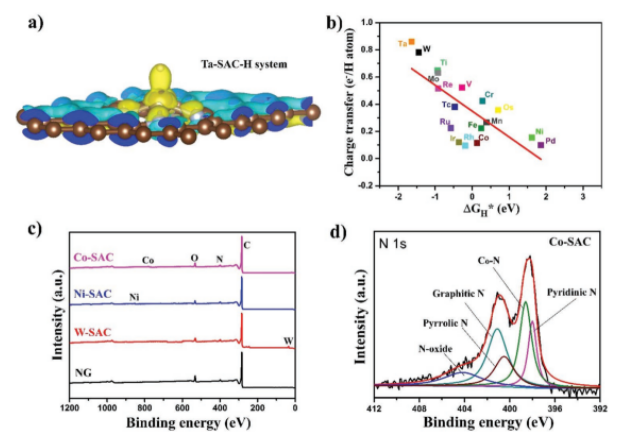
Figure 3. 单原子催化剂的电荷转移和化学组成:a)反应期间从Ta-SAC到氢的电荷转移;b)电荷转移与HER活性(吉布斯自由能)之间的关系;c)Co、Ni和W单原子催化剂与氮掺杂石墨烯(NG)的XPS光谱;d)钴单原子(Co-SAC)的N 1s反褶积光谱。
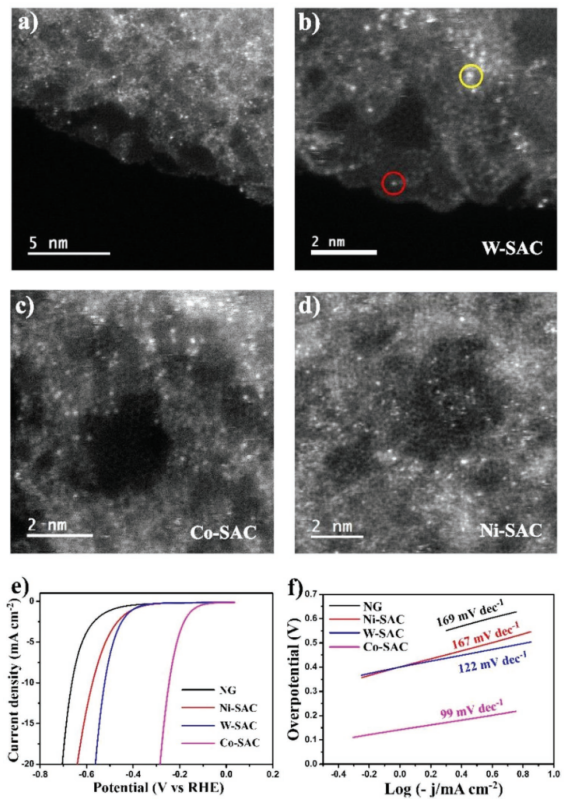
Figure 4. STEM和电化学表征:a)ADF-STEM图像表明孤立的钨单原子均匀分布在整个石墨烯表面;b–d)W-SAC、Co-SAC和Ni-SAC在更高放大倍数下的STEM图像;e)在0.5 M H2SO4中,10 mV/s的扫描速率下对HER的线性扫描伏安曲线;f)相应的Tafel图。
相关研究成果于2019年由香港科技大学Zhengtang Luo课题组,发表在Adv. Energy Mater.(DOI: 10.1002/aenm.201803689)上。原文:Rational Design of Graphene-Supported Single Atom Catalysts for Hydrogen Evolution Reaction。







|
|
| 您的称呼 : | |
| 联系电话 : | |
| 您的邮箱 : | |
| 咨询内容 : | |

|
|