嵌入氮掺杂石墨烯的单原子过渡金属由于其高活性和低材料成本而成为有前途的电催化剂。这些材料已被证明可以催化各种电化学反应,但它们在反应条件下的活性位点仍不为人所知。利用第一原理的密度泛函理论计算,本研究建立了一个依赖于pH值的微观动力学模型,以评估嵌入石墨烯中四倍N-取代的双碳空位的过渡金属催化剂在氧进化反应中的相对性能。本研究发现,在所有的过渡金属上,涉及中间物共同吸附在金属位点上的反应途径是优先的。与纯粹基于热力学的预测相比,这些途径导致了催化活性的增强,并扩大了活性峰值。这些发现证明了研究石墨烯基催化剂和其他二维(2D)材料的反应途径的重要性,这些反应途径涉及由旁观者中间物种装饰的金属活性中心。
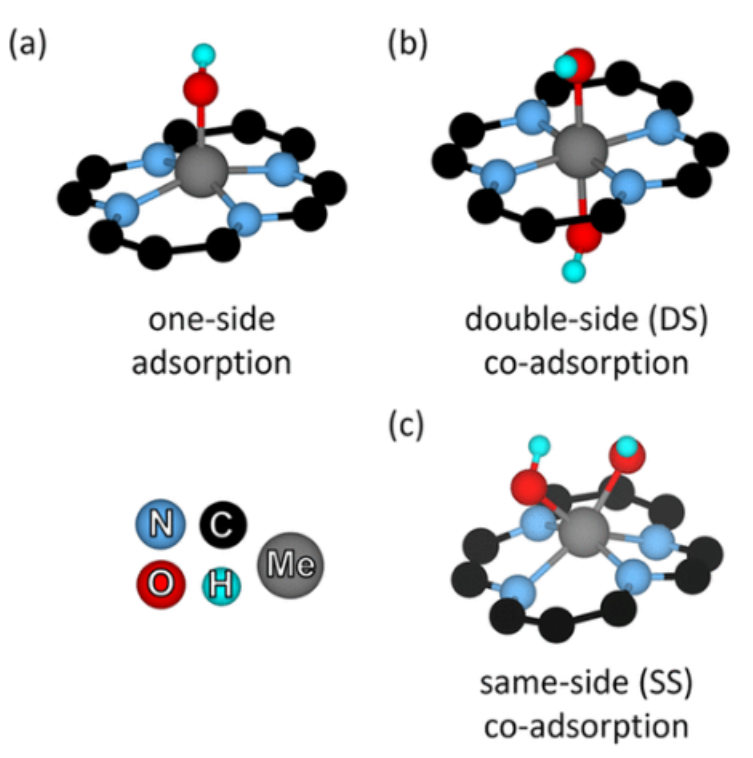
图1. 代表性的结构显示(a)单侧吸附(Me-OH),(b)双侧(DS)共吸附(Me-OH/OH),和(c)同侧(SS)共吸附(Me-OH-OH)。
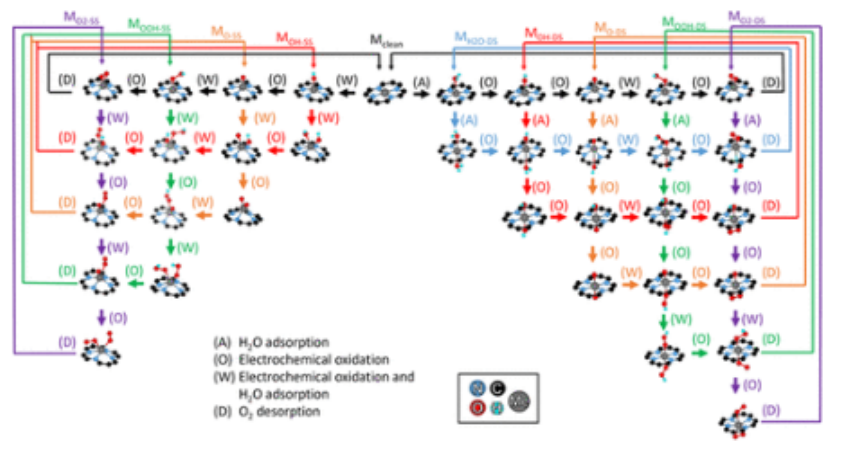
图2. 本工作中考虑的全部OER机制(M
full)。标记的路径对应于机制,其中每个中间物作为一个观察者物种。例如,通过MOH-DS途径,一个单一的OH物种将作为金属中心的旁观者,而反应在石墨烯平面的另一侧进行。在反应网络的任何阶段,金属中心两侧的中间物种被允许进行反应。吸附剂在石墨烯平面同一侧共同吸附的途径用SS表示(例如,M
OH-SS)。吸附物通过双面结构共同吸附的途径,其中吸附物存在于石墨烯平面的两边,用DS表示(例如M
OH-DS)。
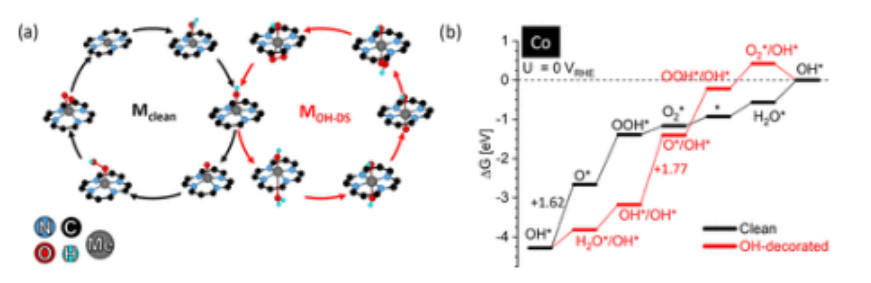
图3:(a)Co
1/G
4-V
2C的活性OER反应途径,包括清洁(M
clean)和双面OH装饰(M
OH-DS)反应机制和(b)相应的自由能图,在U=0 V
RHE和pH值为0时,两种机制都包括最内能基本步骤的反应自由能。

图4. 黑色箭头表示电流反应从清洁机制(M
clean)到完整机制(M
full)的转变。虚线表示从微观动力学模型预测的起始电位(η
onsetMKM),对应于10 mA cm
-2的电流密度和传统热力学预测的M
clean(η
onsetthermo)。覆盖率和DRC曲线是用M
full得出的。对于铁和钌,纯热力学预测估计过电位大于1.0V(表1)。每种金属只有相关状态的覆盖率曲线显示在各自的面板上。通过清洁机制的Ru的电流密度被预测为不活跃,因为它受到O
2解吸的限制。
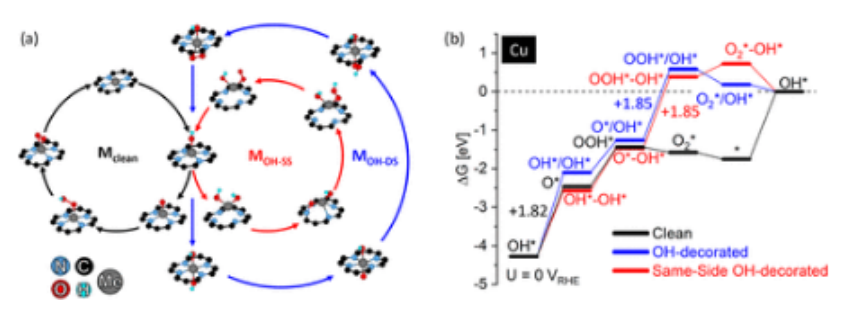
图5.(a) OER反应机理和(b)对于Cu
1/GN
4-V
2C上的清洁(M
clean,黑色)、双面OH修饰(M
OH,蓝色)和同侧OH修饰(M
OH-SS,红色)机理,在U=0V
RHE和pH为0时的相应自由能图。每种机制都包括了最终变基本步骤的反应自由能。
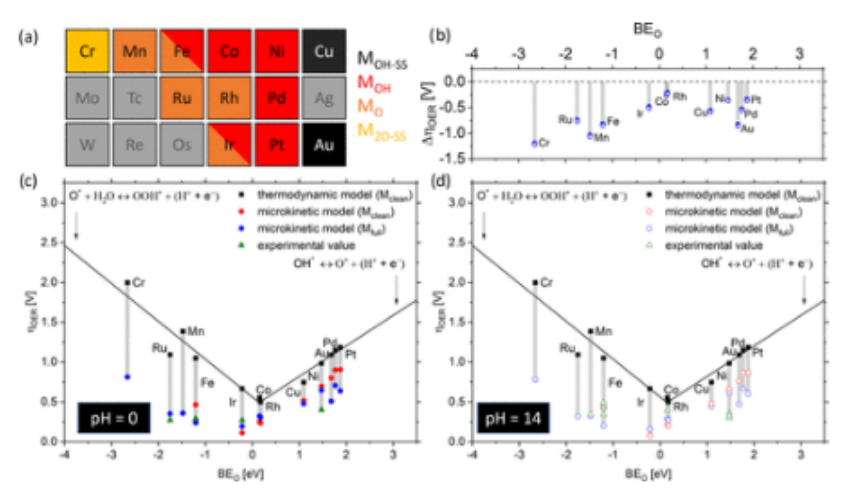
图6.(a)每种金属在起始电位附近的主要反应途径。由多种颜色组成的方框表示不同装饰途径的交叉。灰色方框表示没有考虑的系统,因为它们被预测为不稳定的。(b) 热力学和微观动力学分析预测的过电位的差异(Δη
OER = η
onsetMKM - η
onsetthermo):负值对应于微观动力学分析导致的过电位下降。在pH值为(c)0和(d)14的情况下,单原子过渡金属支持在N掺杂的石墨烯(M
e1/GN
4-V
2C)中的启动电位。热力学分析(黑色方块;线性拟合显示为黑线)和微动能分析(彩色圆圈)的OER起始电位。蓝色和红色圆圈分别对应于M
clean和M
full在10 mA cm
-2下计算的起始电位。不包括Cr、Mn和Ru的红点,因为这些金属被预测为通过清洁机制而不活跃。绿色三角形对应于实验报告的起始电位。
相关研究成果由威斯康星大学麦迪逊分校Manos Mavrikakis等人2023年发表在ACS Catalysis (https://doi.org/10.1021/acscatal.3c00474)上。原文:Insights into the Oxygen Evolution Reaction on Graphene-Based Single-Atom Catalysts from First-Principles-Informed Microkinetic Modeling。
转自《石墨烯研究》公众号










