确定氧化石墨烯(GO)的精确化学结构对于研究其相关性质至关重要。然而,氧化官能团在氧化石墨烯上的分布仍缺乏足够的原子尺度研究。本文基于密度泛函理论进行理论计算,探索氧化石墨烯上羟基和环氧基的热力学分布。影响氧化石墨烯结构稳定性的关键因素有三个:电子分布、位阻和氢键增强,导致羟基和环氧基在氧化石墨烯的两侧紧密分布。因此,我们提出氧化官能团在氧化石墨烯上的选择性近端分布,并认为岛状氧化区更有利于在氧化石墨烯的实际结构上形成。这种选择性的近似前台阶分布模式有助于解释氧化石墨烯表面的起源和演化,并有助于理解二维单层结构的微观表面化学。
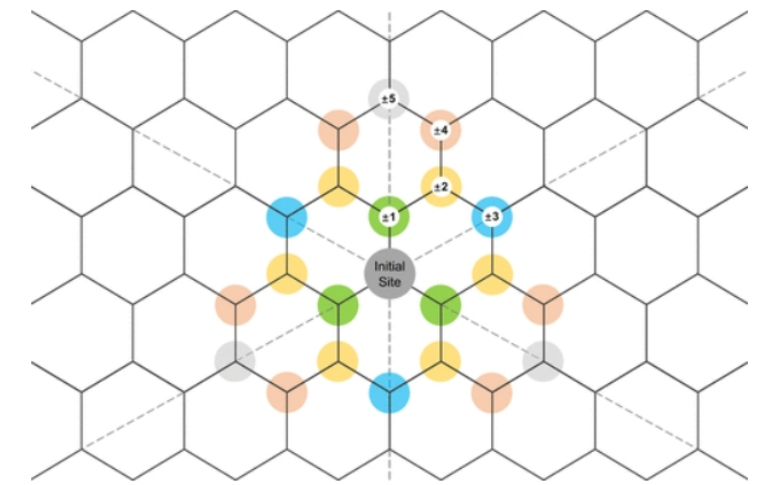
图1. 氧化官能团在氧化石墨烯上的不同分布位点。中间的灰色圆盘表示与原始氧化官能团结合的初始位点,其他不同距离和相对位置的位点用不同颜色和数字标记的圆盘来区分。
正数表示这里的氧化官能团与原氧化官能团分布在氧化石墨烯的同一侧,负数表示这里的氧化官能团与原氧化官能团分布在氧化石墨烯的相反一侧。
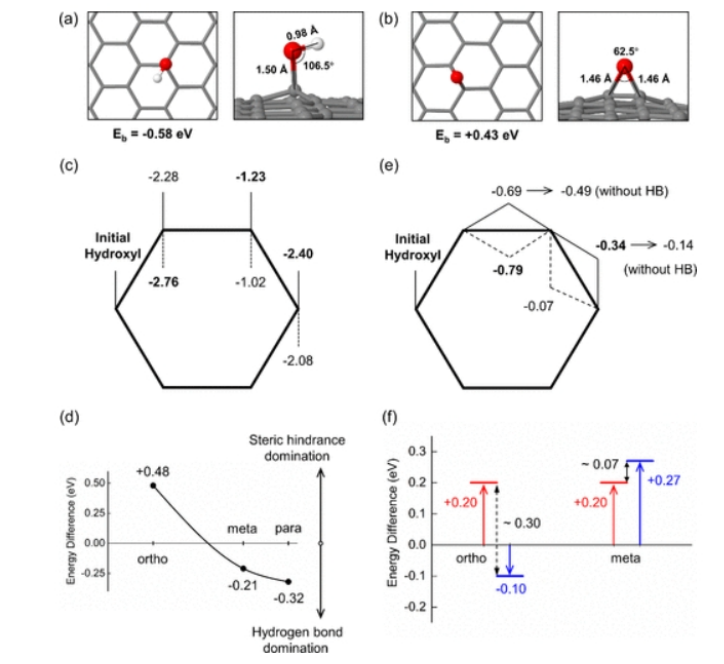
图2: 优化了(a) h
1和(b) e
1的结构和结合能。灰球代表碳原子,红球代表氧原子,白球代表氢原子。(c)不同构型h
2的E
b,其中实线表示表面h
2,虚线表示表面h
2。(d)不同分布部位表面h
2与表面h
2的E
b差异。(e)不同构型下h
1e
1的E
b,实线表示表面h
1e
1,虚线表示下表面h
1e
1。HB表示氢键,后面的值(不带HB)表示表面h
1e
1的E
b,其中O-H不指向环氧基团,不形成氢键。(f)不同分布点h
1e
1的E
b差值,以各分布点地表h
1e
1最稳定的E
b值为基线,设为零。红线表示不同方向上表h
1e
1的E
b差,蓝线表示表前h
1e
1与表h
1e
1的E
b差。
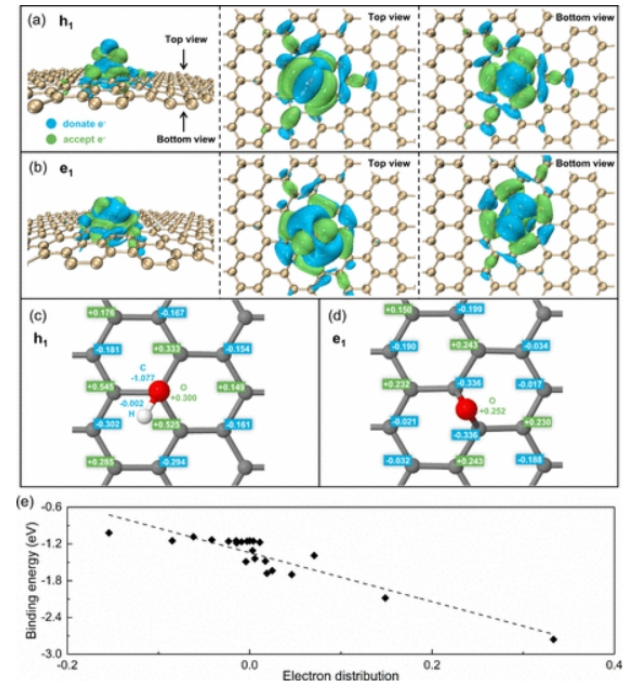
图3. (a) h
1和(b) e
1的电子密度差分析。等值为±0.0005 e
-,其中电子给予量大于0.0005 e
-的区域为蓝色,电子接受量大于0.0005 e
-的区域为绿色。重复(c) h
1和(d) e
1的电荷分析,其中正值表示接受电子,负值表示给予电子。(e)氧化石墨烯表面h
2的结合能与电子分布的关系。
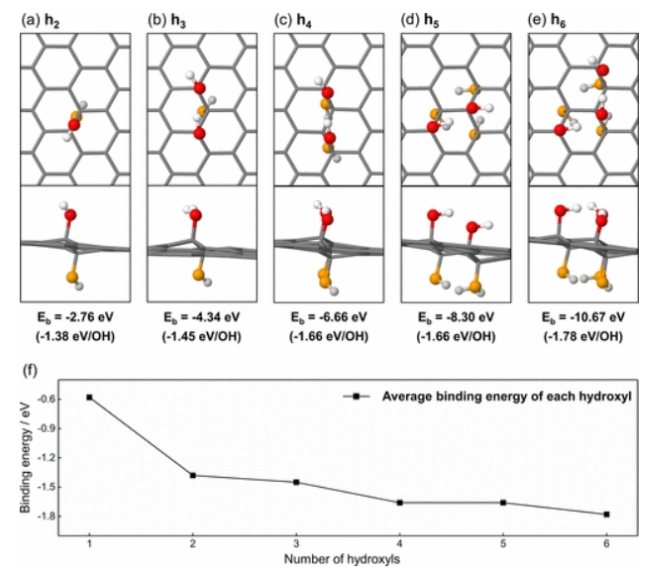
图4. (a) h
2, (b) h
3, (c) h
4, (d) h
5, (e) h
6最稳定的结构和结合能。(f)从h
1到h
6最稳定结构中每个羟基的平均结合能。红色球代表氧化石墨烯平面上方的氧原子,白色球代表氧化石墨烯平面上方的氢原子,橙色球代表氧化石墨烯平面下方的氧原子,浅灰色球代表氧化石墨烯平面下方的氢原子。
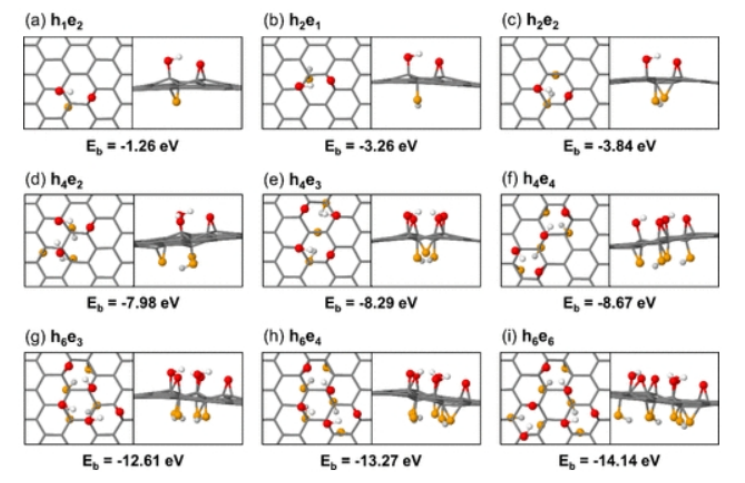
图5.(a) h
2, (b) h
3, (c) h
4, (d) h
5, (e) h
6最稳定的结构和结合能。(f)从h
1到h
6最稳定结构中每个羟基的平均结合能。红色球代表氧化石墨烯平面上方的氧原子,白色球代表氧化石墨烯平面上方的氢原子,橙色球代表氧化石墨烯平面下方的氧原子,浅灰色球代表氧化石墨烯平面下方的氢原子。
相关研究成果由浙江大学
Zhen Xu和
Chao Gao等人2024年发表在The Journal of Physical Chemistry C (链接: https://doi.org/10.1021/acs.jpcc.3c06315)上。原文:Selective Proximate Antarafacial Distribution of Oxidized Functional Groups on Graphene Oxide
转自《石墨烯研究》公众号










