在这项研究中,研究了碳团簇(C
n,其中n的范围从16到26)在MgO表面上的行为。研究考虑了通常在矿石中发现的常见杂质(如Si, Mn, Ca, Fe和Al)掺杂的影响。本研究方法结合了密度泛函理论计算和机器学习力场分子动力学模拟。研究发现,C
21簇簇具有由三个六边形隔离的三个五边形组成的核壳结构,在MgO表面上表现出优异的稳定性,在MgO掺杂表面上表现出“增强结合剂”的作用。分子动力学轨迹表明,与其他尺寸的Cn簇和MgO上的柔性石墨烯层相比,MgO表面稳定的C
21涂层具有更低的迁移率。此外,这种稳定性即使在高达1100K的温度下也能保持。分析了C
n在MgO上的电子定位函数和势函数,发现C
21环中心碳与MgO表面之间存在较高的定位电子密度。这项工作提出了C
21岛可以作为MgO表面的超稳定和低移动的前驱体涂层。这一解释揭示了在石墨烯产品中观察到的实验缺陷,这可以归因于基底上保持冻结和不变的碳岛的迁移率降低。
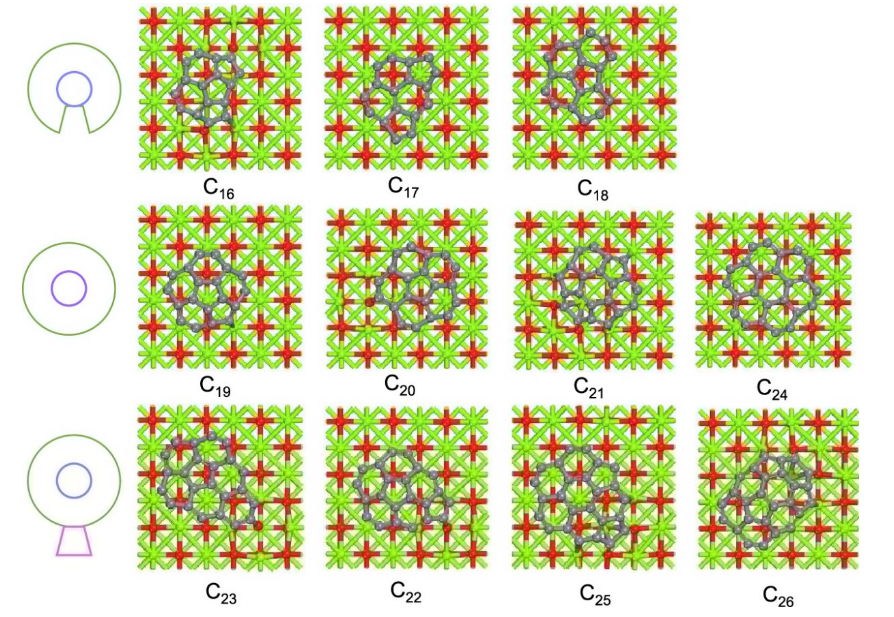
图1. MgO(100)表面C
n (n = 16-26)团簇的基态结构。模型分为三组:C
16-C
18为非封闭核壳结构;C
19-C
21和C
24为闭核壳结构;C
22、C
23、C
25和C
26具有一个或两个附加环(CCS+)的核壳几何结构。
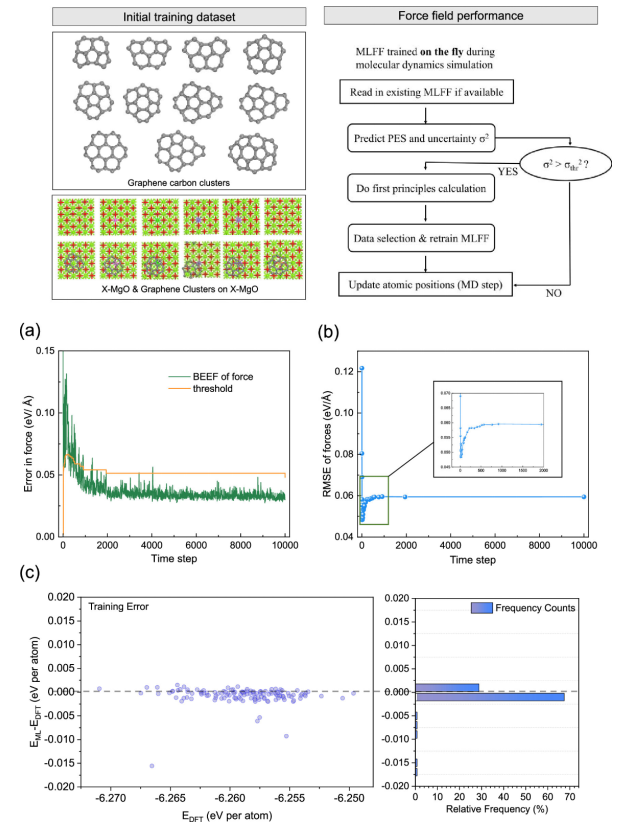
图2. 对C
21@MgO获得的ML-FF进行训练和验证。(a)每个原子的力(BEEF)的贝叶斯误差估计和VASP中动态ML算法为ML- FF的生成设定的阈值标准。(b)相对于DFT结果的力预测的均方根误差(RMSE)。(c) ML-FF MD模拟中132个随机选择结构的能量评估与DFT相比的误差。
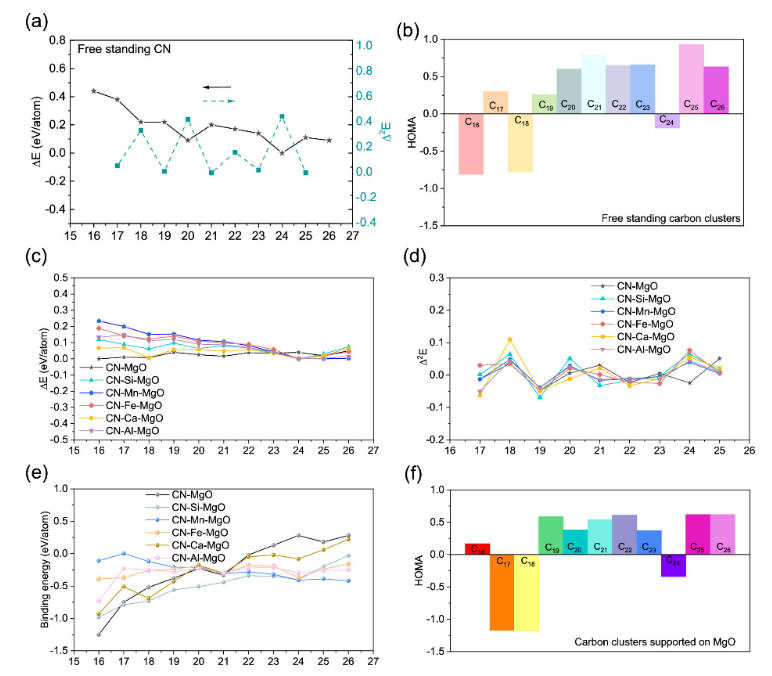
图3. (a)独立碳簇C
n (n = 16-26)及其二阶衍生物(Δ
2E)的结合能(ΔEn)。(b)独立团簇芳香性(HOMA)指数的谐振子模型值。(c)结合能和(d) Cn@MgO (n = 16-26)含杂质和不含杂质的二阶导数。(e) C
n (n = 16-26)在X掺杂MgO (X = Si, Mn, Fe, Ca, Al)上的结合能。(f) Cn@MgO的HOMA指数值(n = 16-26)。使用预训练的ML-FF进行静态优化。
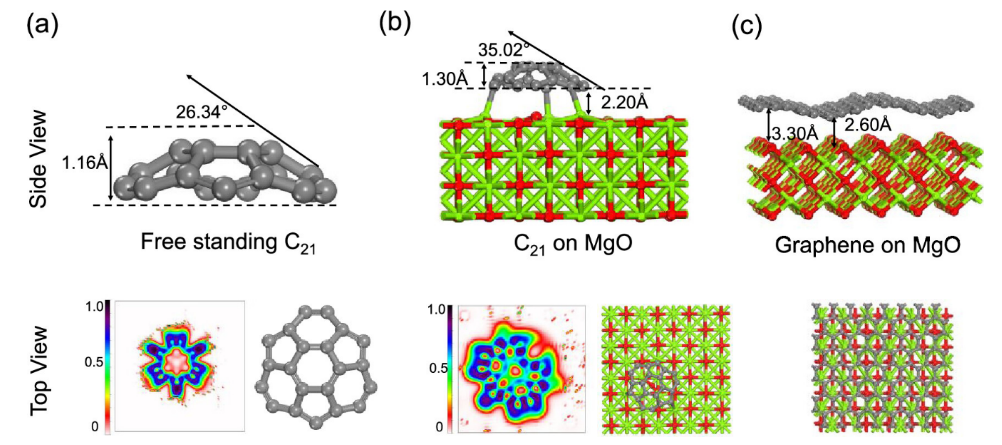
图4. (a)独立C
21的结构特征及其电子定位函数(ELF); (b) C
21@MgO的结构特征及其电子定位函数(ELF)。(c) graphene@MgO的结构特点。使用预训练的ML-FF进行静态优化。

图5. (a) C
21@MgO和(c)石墨烯在MgO (Gra@MgO)上的势能随时间的变化。(b) C
21@MgO和(d) Gra@MgO的ML-FF MD模拟的快照使用OVITO可视化。
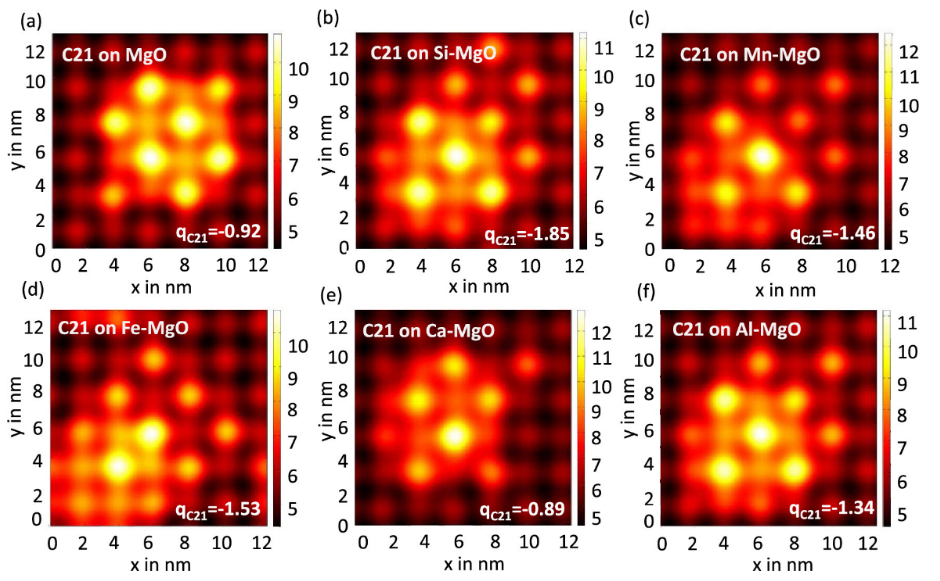
图6. C
21簇在(a) MgO和(b) Si, (c) Mn, (d) Fe, (e) Ca和(f) Al掺杂的MgO上的平均电位。C
21和X-MgO (X = Mg, Si, Mn, Fe和Ca)体系之间的C
21 Bader电荷在右下角标记。
相关研究成果由伦敦大学Rachel Crespo-Otero、伦敦大学玛丽皇后学院Devis Di Tommaso课题组2024年发表在ACS Applied Materials & Interfaces (链接:https://doi.org/10.1021/acsami.4c11398)上。原文:Unveiling Carbon Cluster Coating in Graphene CVD on MgO: Combining Machine Learning Force field and DFT Modeling
转自《石墨烯研究》公众号










